

Research Program Overview

Deep sequencing of normal and tumor tissue showing somatic point mutation in novel RCC tumor suppressor gene BAP1. By Samuel.
Molecular Genetics
A fundamental problem in oncology is heterogeneity (and unpredictability) in the responsiveness of tumors to treatment. This is due, at least in part, to genetic tumor heterogeneity. Thus, it is critical to develop classifications based on the molecular genetics. Using an innovative approach, we identified several novel two-hit tumor suppressor genes and discovered that the BAP1 gene is inactivated in approximately 15% of renal cell carcinomas (RCC) of clear-cell type (ccRCC), the most common type (Peña-Llopis et al., Nat Genet 2012). After VHL and PBRM1, BAP1 is possibly the third most commonly mutated gene in ccRCC. Interestingly, mutations in BAP1 and PBRM1 are largely mutually exclusive (Peña-Llopis et al., Can Res 2013). However, whereas PBRM1 loss is associated with low grade features, BAP1 loss is associated with histological features of aggressive tumors and poor patient survival (Kapur et al., Lancet Oncology 2013). Further analyses in collaboration with investigators at Mayo Clinic have determined that the four subtypes of ccRCC we identified (wild-type, PBRM1-deficient, BAP1-deficient and PBRM1/BAP1 deficient) are associated with different outcomes in patients (Joseph et al., J Urol 2016). The discovery of different biological subtypes of ccRCC may explain the heterogeneity observed in treatment responsiveness and sets the foundation for the development of subtype-specific therapies (Brugarolas J. JCO 2014).
Interestingly, VHL, BAP1 and PBRM1 are all on chromosome 3p, and we speculate that following a mutation in VHL, loss of 3p (which occurs in 80% of ccRCCs) leaves cells vulnerable to the loss of the remaining BAP1 and PBRM1 alleles and subsequent tumor initiation (Brugarolas J. JCO 2014).
Since mutated genes are ultimately responsible for the behavior of cancer cells, our findings have broad implications. In fact, loss of BAP1 sensitizes tumor cells to radiation, an observation that could be exploited for patient treatment (Peña-Llopis et al., Nat Genet 2012). The incorporation of these findings into the clinic will be accelerated by our development of an immunohistochemistry test that can be used in routine medical practice (Peña-Llopis et al., Nat Genet 2012).
We have also discovered a new familial kidney cancer syndrome resulting from germline mutations in BAP1 (Farley et al., Mol Can Res 2013).
In addition, we discovered that the TSC1 gene is mutated in 5% of ccRCCs (Kucejova et al., Mol Can Res 2011). The TSC1 protein negatively regulates mTORC1, and our findings suggest that mutations in TSC1 may predict for sensitivity to mTORC1 inhibitors in the clinic (Brugarolas J. 2012).
More recent studies in collaboration with Genentech have focused on non-clear cell RCC (nccRCC). We reported the first integrated next-generation sequencing analysis of papillary, chromophobe and oncocytomas (Durinck et al., Nat Genet 2015). These studies provide a framework to understand the distinct molecular subtypes of RCC.
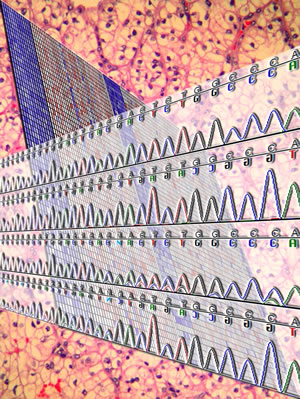
In a background of a ccRCC, a heatmap illustrates the upregulation of hypoxia-inducible genes, including REDD1, which as shown in the chromatogram, is occasionally mutated in ccRCC. Cover from Mol Can Res, vol.9, no.9. Designed by Samuel. For more information, see, Kucejova et al., Mol Cancer Res, 2011.
Pathways
Similarities between two otherwise divergent RCC predisposing syndromes, von Hippel-Lindau, which results from germline mutations in the eponymic gene VHL, and tuberous sclerosis complex, which arises from mutations in the TSC1 and TSC2 genes, led us to hypothesize the existence of a link between pVHL and TSC1/TSC2, which form a complex (Brugarolas et al., Cancer Cell 2003). The VHL gene is inactivated in 80% of ccRCC. VHL encodes the substrate recognition subunit of an E3 ligase complex that targets hypoxia-inducible factor (HIF) for degradation, and VHL inactivation results in constitutive HIF activation. Like pVHL, we found that the TSC1/TSC2 proteins regulated HIF (Brugarolas et al., Cancer Cell 2003). HIF regulation by TSC1/TSC2 is mediated by mTORC1. The TSC1/TSC2 complex negatively regulates mTORC1, and TSC1/TSC2 disruption results in constitutive mTORC1 activation and increased HIF levels and activity. Given the importance of HIF in RCC, the link to mTORC1 presented a rationale for targeting mTORC1 in RCC (Brugarolas et al., New Engl J Med 2007).
mTORC1 plays a critical role in the regulation of cell growth and mTORC1 integrates signals from nutrients, energy stores, growth factors and oxygen. Under unfavorable conditions, mTORC1 is inhibited and cell growth stalls. We previously discovered that mTORC1 inhibition by hypoxia was mediated by the protein regulated in development and DNA damage response 1 (REDD1) (Brugarolas et al., Genes & Dev. 2004). In response to hypoxia, HIF escapes pVHL-mediated degradation and this results in the induction of REDD1, a HIF target gene. REDD1 is both necessary and sufficient for mTORC1 inhibition by hypoxia, and REDD1 action requires an intact TSC1/TSC2 complex (Brugarolas et al., Genes & Dev. 2004).
Because VHL is broadly mutated in ccRCC and HIF is constitutively activated, we expected REDD1 to be overexpressed in ccRCC. This created a paradox, however, as REDD1 induction should inhibit mTORC1, and mTORC1 is frequently activated in ccRCC. In studies of human ccRCC we determined that REDD1 was broadly overexpressed but that this was not associated with mTORC1 inhibition (Kucejova et al., Mol Can Res 2011). REDD1 upregulation was VHL-dependent and mediated by both HIF1 and HIF2. In a tumor-dependent manner, HIF1 and HIF2 were recruited to the REDD1 promoter and were necessary for REDD1 induction. Since we previously showed that REDD1-mediated mTORC1 inhibition requires an intact TSC1/TSC2 complex (Brugarolas et al., Genes & Dev. 2004), we hypothesized that disruption of this complex may explain the activation of mTORC1 despite REDD1 induction. We discovered that the TSC1 gene is somatically mutated in ccRCC, albeit at low frequency (Kucejova et al., Mol Can Res 2011).
REDD1 is not only upregulated in VHL-deficient ccRCC, but also upon Vhl disruption in the mouse. Vhl inactivation in hepatocytes mimics some features of RCC in humans and results in Hif and Redd1 induction. We became interested in trying to understand the role of Hif in this setting. We discovered that acute disruption of Vhl in the mouse liver results in a Hif-mediated inhibition of oxygen utilization and the death of mice within days (Kucejova et al., Oncogene 2011). Vhl disruption blocked fatty acid oxidation as well as ketone and glucose production. Interestingly, simultaneous disruption of Hif abrogated these effects and allowed the mice to survive. As assessed by ex vivo organ perfusion studies and live mice magnetic resonance oximetry, Hif blocked mitochondrial oxygen consumption (Kucejova et al., Oncogene 2011). To our knowledge, this is the first report that Hif is sufficient to block mitochondrial respiration in vivo. Furthermore, the data indicate that, at least in hepatocytes, no other pathways exist to allow oxygen utilization when HIF is active. Should a similar inhibition of mitochondrial respiration be observed in ccRCC, this may open up new opportunities for therapeutic intervention.
To investigate the role of Redd1 in the mouse, we generated a Redd1-deficient mouse strain (Wolff et al., Mol Cell Biol 2011). Using these mice, we discovered an unconventional hypoxia signaling pathway. We established that, while mTORC1 inhibition by hypoxia is mediated by Redd1 in most tissues examined, in hepatocytes, hypoxia signals are transduced in a manner that is independent of Redd1. Furthermore, hypoxia-induced mTORC1 inhibition in hepatocytes is also independent of the Tsc1/Tsc2 complex and Hif. In hepatocytes, oxygen signals are transduced through Lkb1 and AMPK, which, as we showed previously, are dispensable for hypoxia signaling in other cell types (Brugarolas et al., Genes & Dev. 2004).
REDD1 is believed to act by sequestering 14-3-3 proteins away from TSC2 thereby activating TSC2 and inhibiting mTORC1. However, the 14-3-3 binding site is not evolutionarily conserved and how REDD1 would sequester 14-3-3 proteins, which are very abundant, was unclear. To evaluate this model, we performed structural studies of REDD1. In collaboration with the Zhang lab, we determined the crystal structure of REDD1 at 2.0 Å resolution (Vega-Rubin-de-Celis et al., Biochem 2010). We found that the putative 14-3-3 binding motif does not conform to any 14-3-3 binding motifs known and 14-3-3 proteins could not be docked on this site. Furthermore, mutation of residues thought to be critical for 14-3-3 binding, including the putative phosphoacceptor serine, had no appreciable effect on REDD1 function. Altogether these data strongly suggest that the accepted model of REDD1 action is probably incorrect. In addition, by surface conservation mapping and mutagenesis studies we identified a patch, likely to be involved in protein-protein interactions, that was required for REDD1-mediated inhibition.
Interestingly, REDD1 is not only implicated in hypoxia-induced mTORC1 inhibition, but the cell also uses REDD1 to inhibit mTORC1 in response to viral infections (Mata et al., Nat Chem Biol 2011). By inducing REDD1 and inhibiting mTORC1, cells reduce viral protein replication and protect themselves.
Finally, we have discovered a novel effector of mTORC1, the transcription factor EB (TFEB), an oncogenic transcription factor translocated in RCC (Pena-Llopis et al., EMBO J 2011). TFEB is a master regulator of lysosomal genes and this work links mTORC1 to lysosome biogenesis.
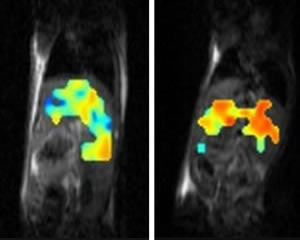
MRI oximetry showing blockade of mitochondrial respiration by HIF in the liver of a mouse with consequent pO2 upregulation. Normal liver shown in the left for comparison. Adapted from Kucejova et al., Oncogene 2011.
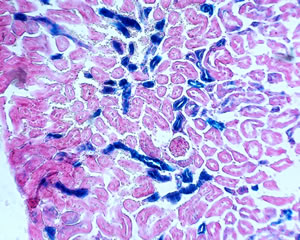
Redd1 expression in the kidney of a genetically engineered Redd1 gene-trap mouse. Adapted from Wolff et al., Mol Cell Biol, 2011.
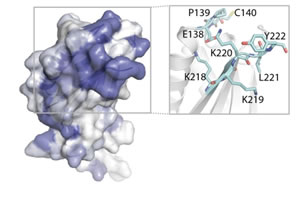
Structure of the REDD1 protein. Adapted from Vega-Rubin de Celis et al., Biochemistry, 2010.
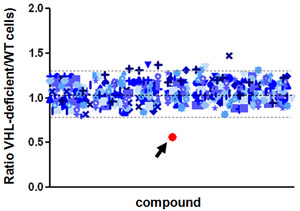
A chemical (red) that selectively kills kidney cancer cells deficient in VHL. By Nick.
Drug Identification
Genes mutated in RCC form the basis of chemical screens for drug discovery. We have developed an assay for high-throughput evaluation of compounds with synthetic lethal activity against any gene mutated in renal cancer. We recently completed a 200,000 compound screen with an innovative design and have identified several chemical leads that selectively target genetic abnormalities in ccRCC tumor cells. Our next goal is to evaluate the activity of emerging compounds in the animal tumor model we developed.
In addition, we have found that a drug used for the treatment of leukemia, omacetaxine (formerly homoharringtonine) has activity against a subset of VHL-deficient ccRCCs (Wolff et al., Oncotarget 2015).
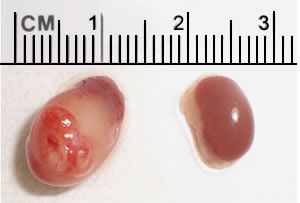
Human renal cancer in a mouse kidney (left). By comparison, a normal mouse kidney is shown on the right. By Sharanya and Toshi.
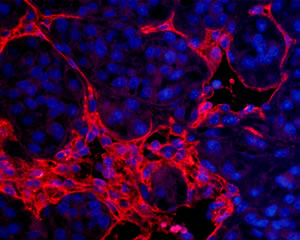
Blood vessels (CD31 staining) in a human tumor growing in a mouse kidney. By Anh.
Tumor Modeling
We have generated the first mouse model of RCC that reproduces the treatment responsiveness of patient tumors. We determined that tumors from patients when implanted orthotopically in mice (tumorgrafts) retain the histology, gene expression, and the vast majority of the mutations of the corresponding patient tumor. To date, we have implanted tumors from over 700 patients. Most importantly, we have established that tumorgrafts in mice reproduce the drug responsiveness of RCC in patients (Sivanand et al., Sci Transl Med 2012). We have developed protocols for the use of these mice in preclinical drug testing (Pavia-Jimenez et al., Nat Protoc 2014). Our goal is to use these mice to evaluate compounds emerging from our drug screens. Tumorgraft-bearing mice are also being used in co-clinical trials where both the patient and the corresponding mouse are being treated with a novel agent. Video.
Transplantation tumor models are being complemented by genetically-engineered mouse models, where we are able to study the effects of particular kidney cancer genes in isolation (Wang et al., PNAS 2014; Gu et al., Canc Discov 2017).
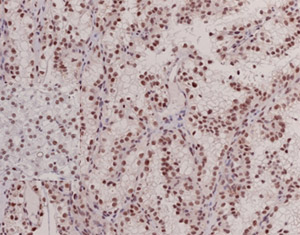
Immunohistochemistry test for BAP1 protein. By Payal.
Biomarkers
The application of discoveries about the biology of tumors to the clinic is accelerated by the development of biomarkers. Biomarkers are readouts of tumors. Our molecular genetics program led to the discovery of a novel tumor suppressor implicated in renal cancer, BAP1, and we have developed a biomarker that can readily be incorporated into routine clinical practice to determine the status of BAP1 in patient tumors (Peña-Llopis et al., Nat Genet 2012).
Our efforts are also focused on the identification of biomarkers that can detect tumors most likely to respond to particular therapies. We are evaluating both inhibitors of angiogenesis and mTORC1 inhibitors. Inhibitors of angiogenesis are particularly challenging because the drugs do not target the tumor directly, but rather, they target blood vessels. However, we have developed an innovative approach that allows us to study the effects of drugs in the crosstalk between tumor and blood vessel cells (Tran et al., MCB 2016).
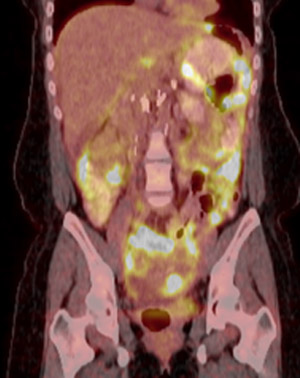
PET/CT of a patient with peritoneal carcinomatosis from an FH-deficient RCC that was subsequently treated with 2DG. For more information, see Yamasaki et al., Nat Rev Urol, 2011.
Clinical Trials
Our aim is to identify compounds from our chemical screens that show activity in our mouse model and which can ultimately be developed into drugs and tested in clinical trials. This process will likely involve partnerships with the pharmaceutical industry. We are also interested in understanding how tumors become resistant to current drugs. In fact, a clinical trial has been opened to explore the mechanism by which tumors develop resistance to mTORC1 inhibitors. Insight into how tumors become refractory to mTORC1 inhibitors may lead to more effective ways to block this pathway and inhibit tumor growth.
In collaboration with Peloton Therapeutics, Inc., we recently evaluated a first-in-class inhibitor of arguably the most important driver of ccRCC, HIF-2 (Wang et al., Nature 2016, Courtney et al., J Clin Oncol, 2018). Video. Other concepts being evaluated include whether stereotactic radiation increases the probability of tumor responses to high-dose IL2 and we are involved in different trials with immune checkpoint inhibitors. A complete list of clinical trials can be found here.
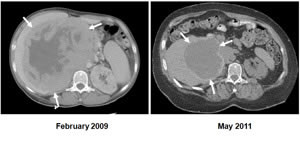
First report that mTORC1 inhibitors may be effective against renal epithelioid angiomyolipomas in a patient who had been informed that his cancer was terminal. For more information, see Wolff et al., J Clin Oncol, 2010.
Personalized Medicine
We strive to provide patients with a tailored approach to treatment. Our six research strands provide a conceptual framework for a “bench-to-bedside” pipeline in which fundamental biology supports clinical innovation. For examples of our translational approach, see Straka et al., J Clin Oncol 2013, Jacobs et al., J Clin Oncol 2013, Yamasaki et al., Nat Rev Urol 2011, Wolff et al., J Clin Oncol 2010, Brugarolas et al., J Clin Oncol 2008.